

Mycobacterial Evolution, Physiology and Virulence
We investigate the remarkable strategies that Mycobacterium tuberculosis (Mtb) has evolved to survive and thrive within the hostile environment of the human host. By studying the evolution, physiology, and virulence of this pathogen, we aim to uncover vulnerabilities that can be exploited for new therapeutic interventions.
Mycobacterial Response to Host-Imposed Stress
Principal investigators | Pierre DUPUY, Claude GUTIERREZ, Florence LEVILLAIN, Yannick POQUET
Other personnel | Louis BENASTRE
Mtb’s unique evolutionary history, shaped by ancient horizontal gene transfer events, has endowed it with a repertoire of specialized systems that allow it to sense, respond to, and withstand a wide range of environmental stresses and host immune defenses. Among these, toxin-antitoxin (TA) modules, including RES-Xre and MenT/MenA, play a pivotal role in modulating bacterial physiology. These systems enable Mtb to finely regulate metabolic activity, selectively inhibit protein synthesis, and enter a transiently dormant state that enhances survival during periods of stress, including nutrient limitation, oxidative bursts, and phage predation.
Our laboratory has characterized several of these TA systems to understand the molecular triggers that activate toxins, the regulatory circuits that control antitoxin expression, and the downstream effects on bacterial physiology. By combining genetic, biochemical, and structural approaches, we aim to map the functional networks of these modules and uncover how they contribute to Mtb’s resilience. Beyond their fundamental biological importance, TA systems also represent highly promising targets for next-generation anti-TB therapies, as their inhibition could disrupt bacterial stress responses and sensitize Mtb to existing drugs. Through this work, we hope to provide new insights into bacterial survival strategies while paving the way for innovative approaches to combat TB.
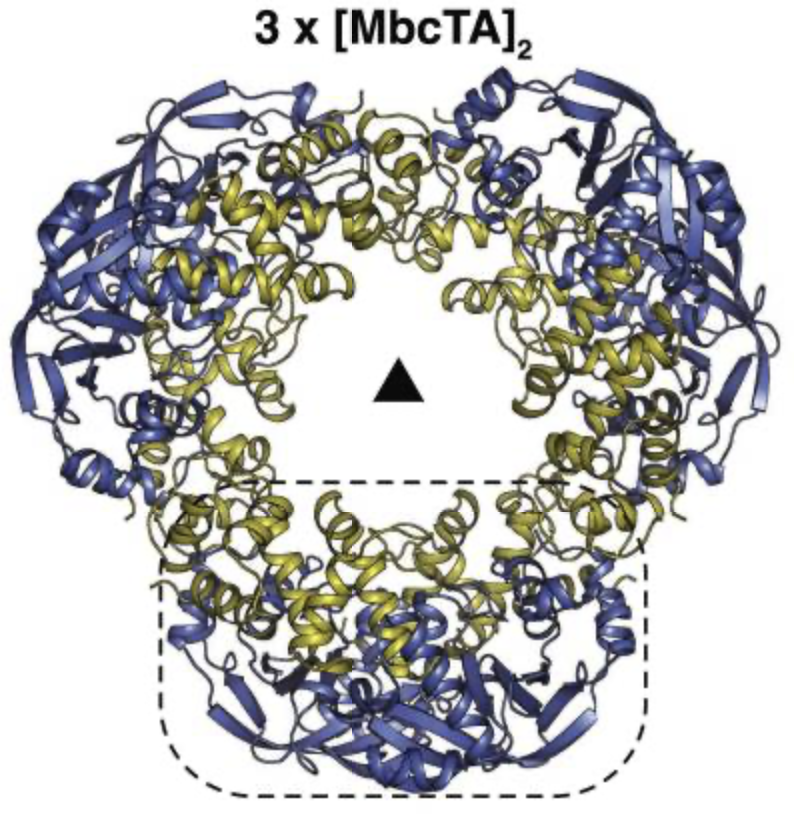
Overall structure of the MbcTA heterododecamer consisting of three heterotetramers (3x[MbcT-MbcA]2) arranged around a 3-fold symmetry axis (as indicated by a black triangle). The dashed line box in the front view (left) represents one [MbcT-MbcA]2 heterotetramer formed by two MbcT (blue) and two MbcA (yellow) molecules as indicated in the side view (right). From Freire*, Gutierrez* et al. (2019) Mol Cell.
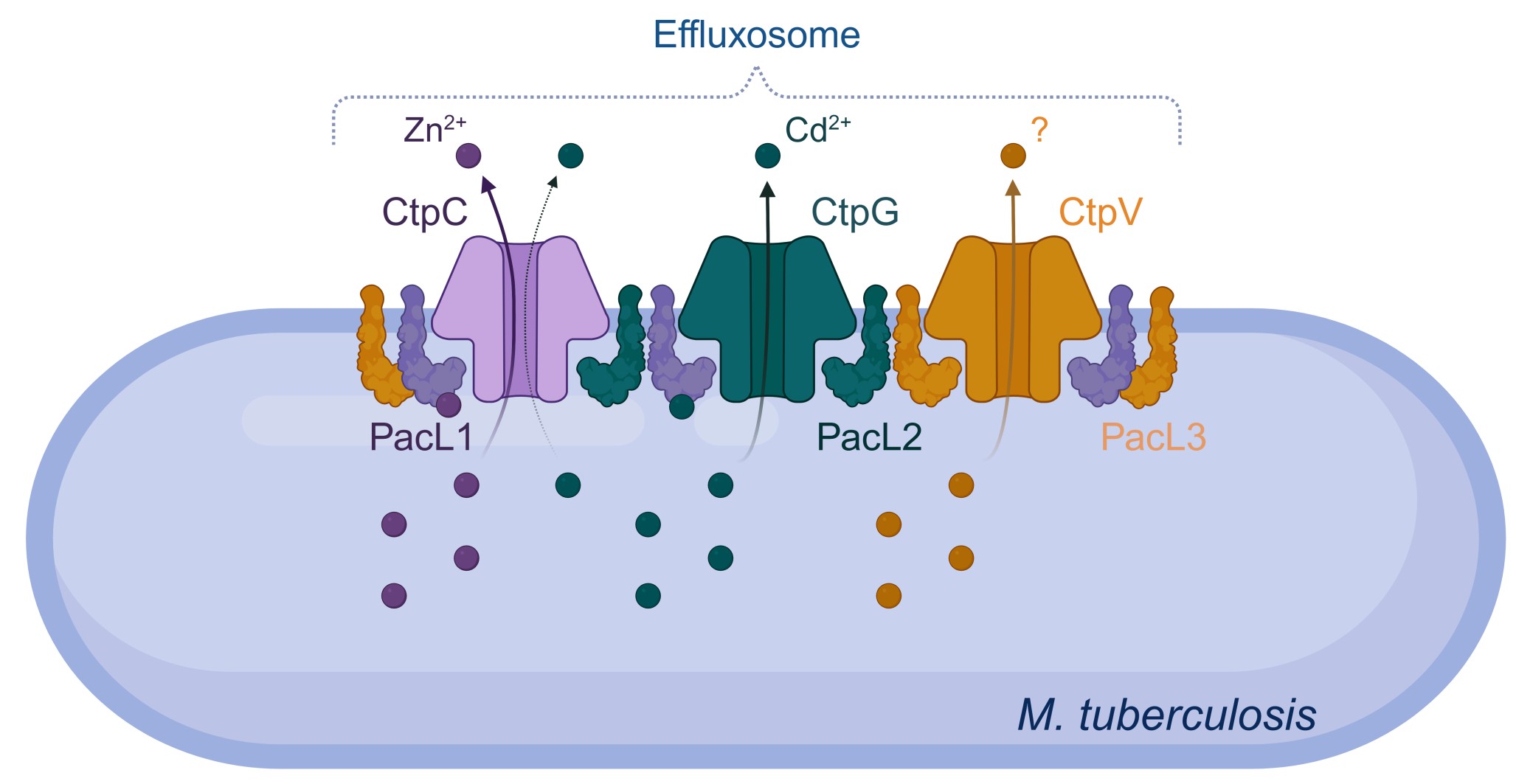
Conceptual model of mycobacterial effluxosome for multi-metal cross-resistance. The P-ATPases CtpC and CtpG facilitate the efflux of zinc and cadmium from the cell, while the role of CtpV remains unclear. The PacL1, PacL2, and PacL3 proteins interact with each other through the GXXG motif in their transmembrane domains and with the metal-binding N-terminal domains of CtpC, CtpG, and CtpV via EA repeat sequences in their cytoplasmic domains. These interactions are essential for the metal resistance mediated by these P-ATPases and for clustering them into dynamic mobile membrane complexes. PacL1, but not PacL2, binds a variety of metal ions, thereby enhancing the metal tolerance conferred by the P-ATPases. From Dupuy et al. (2026) EMBO J.
Our lab has uncovered how metal stress shapes the host–pathogen arms race during Mtb infection. We first showed that macrophages use zinc intoxication as an antimicrobial strategy, with zinc accumulating in Mtb-containing phagosomes. Parallel transcriptional profiling revealed coordinated heavy metal stress responses in both host and pathogen, including induction of host metallothioneins and ZnT1, and upregulation of mycobacterial P1B-type ATPases such as CtpC, CtpG, and CtpV.
We demonstrated that CtpC is essential for resistance to zinc poisoning, as its loss leads to intracellular zinc accumulation and impaired bacterial growth. We then identified PacL1 as a zinc-binding, chaperone-like partner that stabilizes CtpC at the plasma membrane; its absence destabilizes CtpC and sensitizes bacteria to zinc stress. More recently, we described a family of PacL proteins (PacL1–3) that organize P-type ATPases into dynamic membrane platforms we termed “effluxosomes,” coordinating resistance to zinc, cadmium, and copper through spatially structured multi-protein assemblies.
Together, our work establishes metal homeostasis as a central determinant of Mtb persistence and a promising target for innovative antimicrobial strategies.
We have been dissecting the metabolic strategies that Mtb uses to survive in the nutrient-limited and hostile host environment. Early on, we identified AnsP1 as the unique aspartate transporter in Mtb. Metabolomic analysis of an AnsP1 mutant showed that aspartate is a primary nitrogen source essential for host colonization, as its loss markedly reduces virulence. Building on this, we discovered that Mtb uses the asparagine transporter AnsP2 and the secreted asparaginase AnsA to acquire nitrogen and resist phagosomal acid stress. While AnsP2 can be partially compensated by other transporters, AnsA is indispensable for preventing phagosome acidification and supporting intracellular growth; mutants lacking AnsA are severely attenuated in macrophages and mice, linking metabolic adaptation directly to virulence.
We also examined horizontally acquired genes contributing to pathogenicity, focusing on the moaA1-D1 cluster involved in molybdenum cofactor biosynthesis. Unlike their paralogs, moaA1-D1 is strongly induced under hypoxia. Loss of these genes impairs nitrate respiration and survival under oxygen-limiting conditions in vitro, while their expression in Mycobacterium kansasii enhances hypoxic survival. In vivo, moaA1-D1 deletion reduces persistence in hypoxic granulomas, highlighting a pathway essential for resisting host-imposed hypoxia.
More recently, we extended our investigation to sulfur metabolism, a critical but understudied aspect of Mtb physiology. Sulfur fuels essential pathways, including redox buffering and coenzyme production, yet the primary sulfur sources exploited during infection were unclear. We discovered that Mtb acquires inorganic sulfate via the SubI-CysTWA transporter during macrophage infection. High-resolution nanoSIMS analysis revealed significant incorporation of sulfate-derived ³³S in intracellular bacteria, correlating with metabolic activity. Deletion of subI abolished sulfate uptake, impairing growth in vitro and reducing bacterial survival in macrophages and infected murine lungs. These findings demonstrate that, unlike many intracellular pathogens, Mtb relies on an energetically costly inorganic sulfate assimilation pathway to sustain redox balance and resist nitrosative stress. Given that mammalian cells lack this pathway, SubI-CysTWA represents a pathogen-specific vulnerability that could be exploited therapeutically, either to directly inhibit Mtb survival or to sensitize the bacterium to oxidative stress induced by antibiotics.
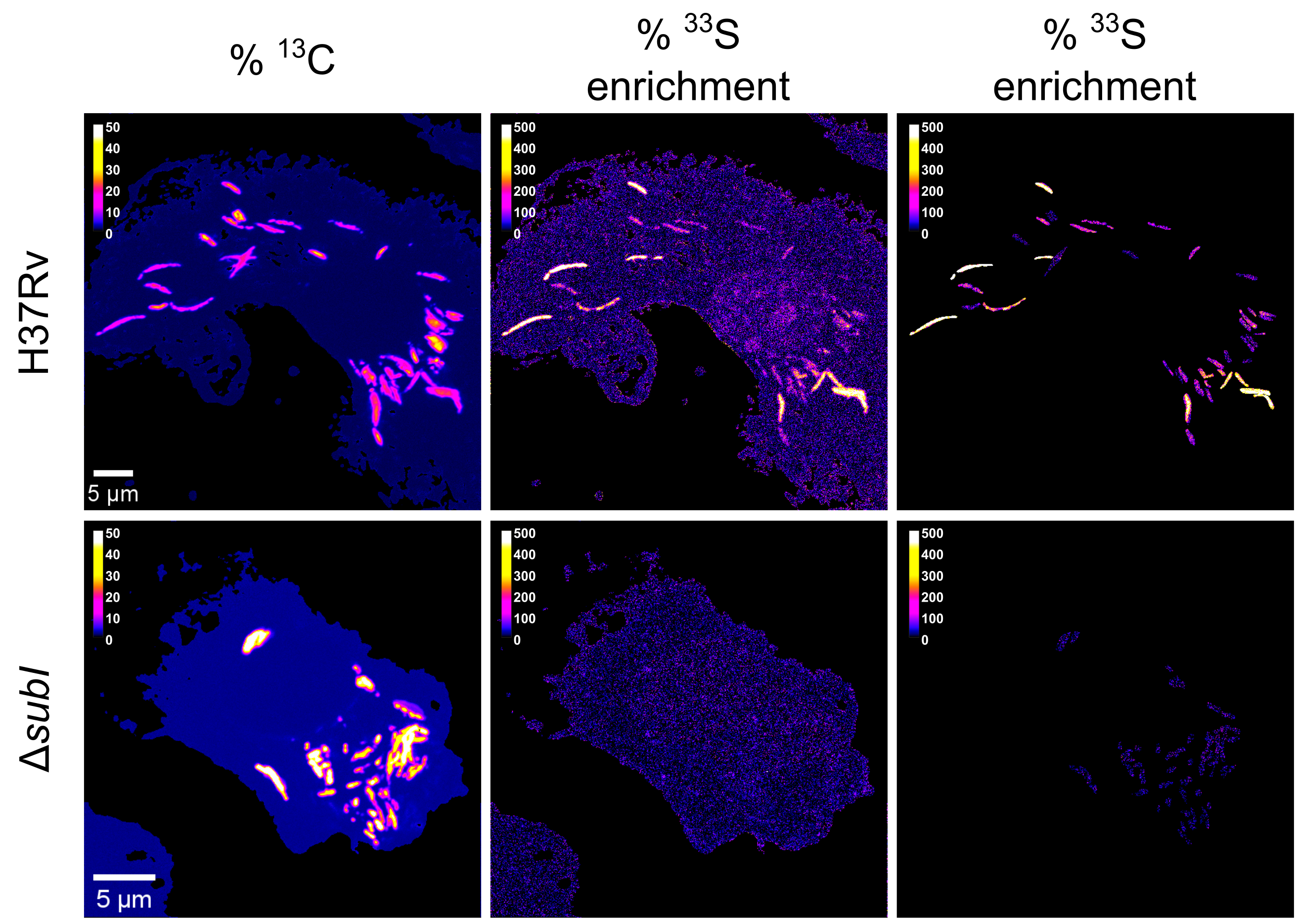
SubI-CysTWA mediates sulfate import in intracellular M. tuberculosis. Murine bone-marrow derived macrophages were infected with 13C-labeled Mtb H37Rv or ∆subI in presence of 33S-sulfate (2 mM) and 15N-cystine (0.2 mM). Three days post-infection, samples were fixed and 13C, 33S and 15N isotopic enrichments were measured by nanoSIMS. NanoSIMS images of representative macrophages infected with wild-type or ∆subI bacteria. From Le Mouëllic et al. (2025) PNAS
Collectively, our work shows that Mtb’s metabolic adaptations are tightly linked to virulence and persistence. By mapping nitrogen and sulfur acquisition and hypoxia-resistance pathways, we reveal the biochemical strategies that allow the bacterium to thrive in the host and identify multiple pathogen-specific targets for new anti-TB therapies.
A nascent project now explores how Mtb responds to host-induced stress, focusing on DNA damage, repair, and genome stability. We aim to understand how DNA breaks arise during infection, how repair systems coordinate with metabolism and stress responses, and how these processes support persistence, virulence, and antibiotic tolerance. Using molecular genetics, infection models, and advanced analytics, this work seeks to uncover vulnerabilities that could inform novel therapeutic strategies.
Mycobacterial Exometabolome and Virulence
Principal investigators | Fabien LETISSE, Philippe VOGELEER
Other personnel | Saurabh CHUGH, Tejan LODHIYA
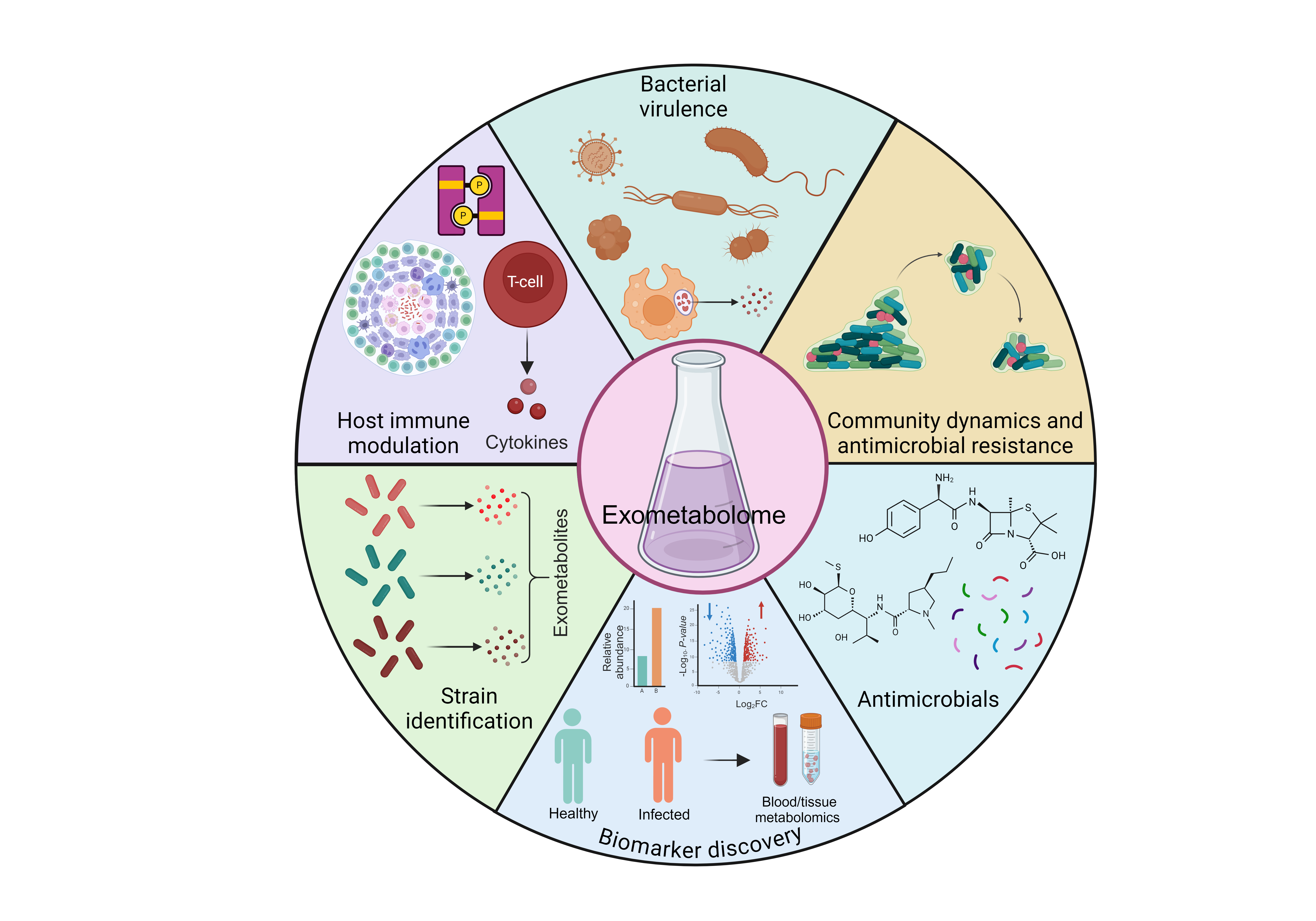
Overview of the potential applications of the microbial exometabolome. Microbial exometabolome research holds potential in (i) exploring bacterial virulence mechanisms, (ii) studying community dynamics and antimicrobial resistance, (iii) modulating host-immune interactions, (iv) strain identification, (v) biomarker discovery, and (vi) the identification of novel antimicrobial compounds. The molecular structures shown are representative examples. The figure was created using BioRender. From Chugh et al. (2025) Trends Microbiol
Beyond intracellular metabolism, our research increasingly interrogates the Mtb exometabolome—the spectrum of metabolites secreted into the extracellular milieu. These molecules are not mere byproducts; they function as dynamic effectors that can reprogram host cell physiology, modulate immune signaling, and shape inflammatory landscapes, effectively serving as underrecognized virulence determinants. Leveraging high-resolution mass spectrometry and NMR-based metabolomics, we aim to systematically map and characterize this exometabolome, revealing how Mtb exploits metabolic plasticity to manipulate the host environment, promote survival, and resist therapeutic interventions, thereby uncovering novel metabolic vulnerabilities for targeted therapies.
By combining genetic, biochemical, and imaging approaches, our team continues to dissect the molecular networks that enable Mtb to persist, evade host defenses, and cause disease. Our research integrates insights into metabolic adaptation, stress resistance, and host-pathogen interactions, providing a foundation for the development of novel drugs and vaccines aimed at controlling TB more effectively. Through these studies, we aim not only to deepen our understanding of Mtb biology but also to identify strategies that tip the balance in favor of the host, offering new hope in the fight against one of humanity’s oldest and most persistent infectious diseases.