

Immunity to TB & TB-HIV Co-Infection
Our laboratory investigates how Mycobacterium tuberculosis (Mtb) interacts with the human immune system and how these interactions are altered during TB-HIV co-infection. Understanding these processes is critical for identifying new strategies to enhance immunity, limit inflammation, and improve disease outcomes.
Glycans, Lectins and Immunity to Tuberculosis
Principal Investigator | Yoann ROMBOUTS
Other Personnel | Maxime KAISER, Stella ROUSSET
Our laboratory has made sustained contributions to understanding how C-type lectins and glycans regulate immunity to Mtb, progressively uncovering key mechanisms of pathogen recognition and immune modulation in TB.
Our early work demonstrated that DC-SIGN (CD209), a C-type lectin expressed by human dendritic cells, is a major receptor mediating Mtb entry through recognition of mannose-rich mycobacterial glycoconjugates such as lipoarabinomannan (LAM). We further showed that DC-SIGN is expressed in lung dendritic cells and alveolar macrophages from TB patients, where it marks preferential target cells for Mtb in vivo. Extending these findings to experimental models, we identified the murine DC-SIGN homologue SIGNR3 as a nonredundant protective receptor required for optimal inflammatory cytokine production and resistance to pulmonary TB.
Our work then turned to inhibitory C-type lectins, particularly DCIR (dendritic cell immunoreceptor). We showed that DCIR plays a critical role in calibrating lung inflammation during TB by sustaining type I interferon signaling in dendritic cells, thereby balancing bacterial control and immunopathology. More recently, we identified LRP1 as the long-sought endogenous ligand of DCIR and defined the structural basis of DCIR-glycan recognition. This work uncovered a novel glyco-immune regulatory axis in which DCIR, through recognition of specific complex N-glycans, modulates signaling downstream of ITAM-associated receptors such as Dectin-1 and FcγRs, refining our understanding of how activating and inhibitory lectins integrate signals during infection. The molecular mechanisms underlying DCIR signaling are currently being elucidated, and tools to modulate DCIR activity in TB and other inflammatory settings are being developed.
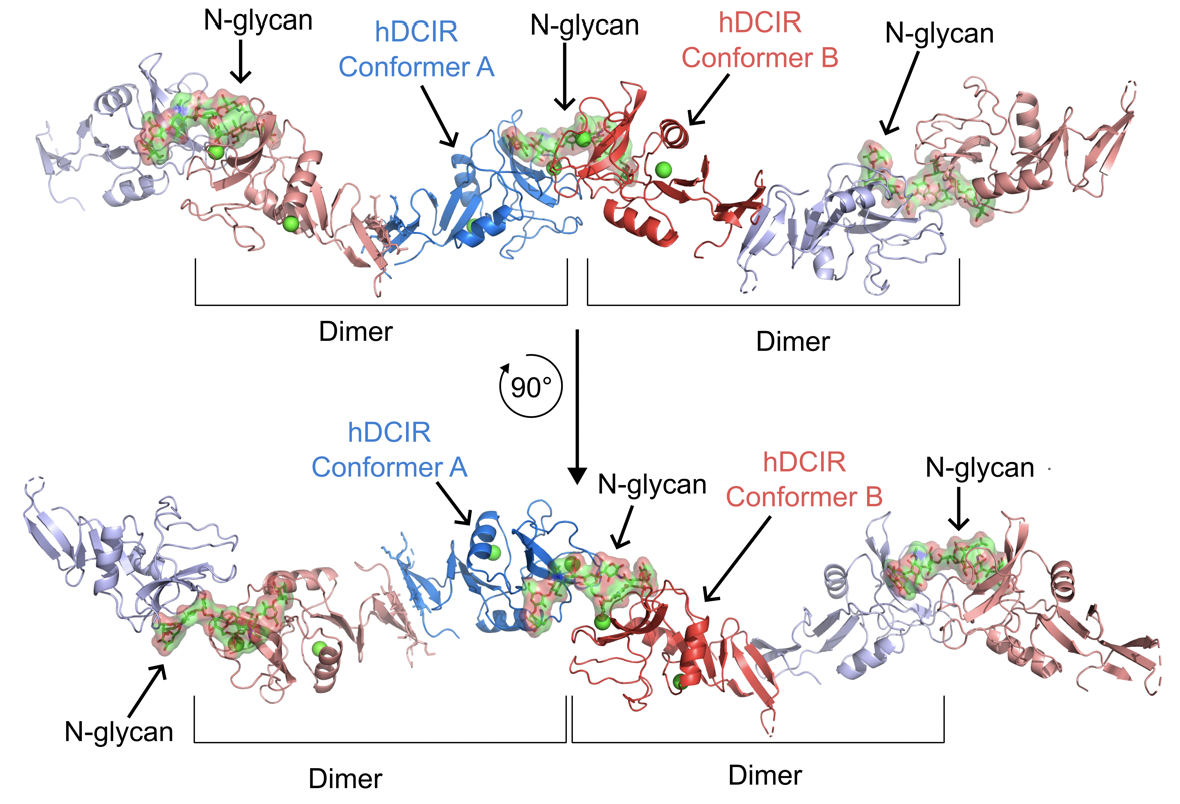
Structural analysis reveals the dimeric organization of hDCIR. Crystal packing of the hDCIR-ECD/biantennary complex-type N-glycan complex. Protein, carbohydrate, and calcium ions are shown in ribbon (blue and red for conformer A and B respectively), stick (green/red), and sphere (light green) models, respectively. The electron density of the N-glycan is represented by the grey mesh. From Raymond*, Sneperger*, Rousset* et al. (2025) Research Square
Beyond lectin receptors, we have investigated how glycan modifications themselves shape immunity. Most recently, we demonstrated that ST8SIA4-dependent polysialylation is a critical regulator of inflammatory monocyte trafficking. Using genetic models, we showed that polysialylation controls CCR2-dependent monocyte egress from the bone marrow at steady state and during Mtb infection by regulating receptor organization, cytoskeletal dynamics, and chemokine responsiveness. These findings reveal glycosylation as an essential determinant of immune cell mobilization and host defense.
Together, our work establishes C-type lectins and glycan modifications as central regulators of innate immune recognition, inflammation, and cell trafficking in tuberculosis. By dissecting both receptor-mediated glycan sensing and glycan-dependent immune cell regulation, we aim to identify new molecular targets to fine-tune host immunity and improve TB outcomes.
Lymphoid Cells in Immunity to TB
Principal Investigator | Denis HUDRISIER
Other Personnel | Annie BEHAR, Maxime CAOUAILLE
Our research investigates how diverse immune cell populations are reprogrammed within the lung during tuberculosis and how these interactions determine protection or pathology. Early on, we uncovered an unexpected antibody-independent role for B cells during Mtb infection, demonstrating that Mtb-stimulated B cells produce type I interferon through a STING-dependent pathway. This B cell–derived type I IFN promotes the polarization of macrophages toward a regulatory/anti-inflammatory phenotype, revealing a previously unrecognized mechanism by which B cells shape the pulmonary immune environment in both mice and patients with TB.
Building on this foundation, we showed that tissue-resident innate lymphoid cells (ILCs) are highly plastic during infection, with IL-18Rα⁺ ILCs differentiating into protective, interferon-γ–producing ILC1-like cells under the influence of type 1 cytokines and a glycolytic metabolic program. BCG-induced type 1 inflammation further enhances the early generation of these protective ILC1-like cells upon secondary Mtb challenge, highlighting how local inflammatory cues tailor innate lymphocyte function.
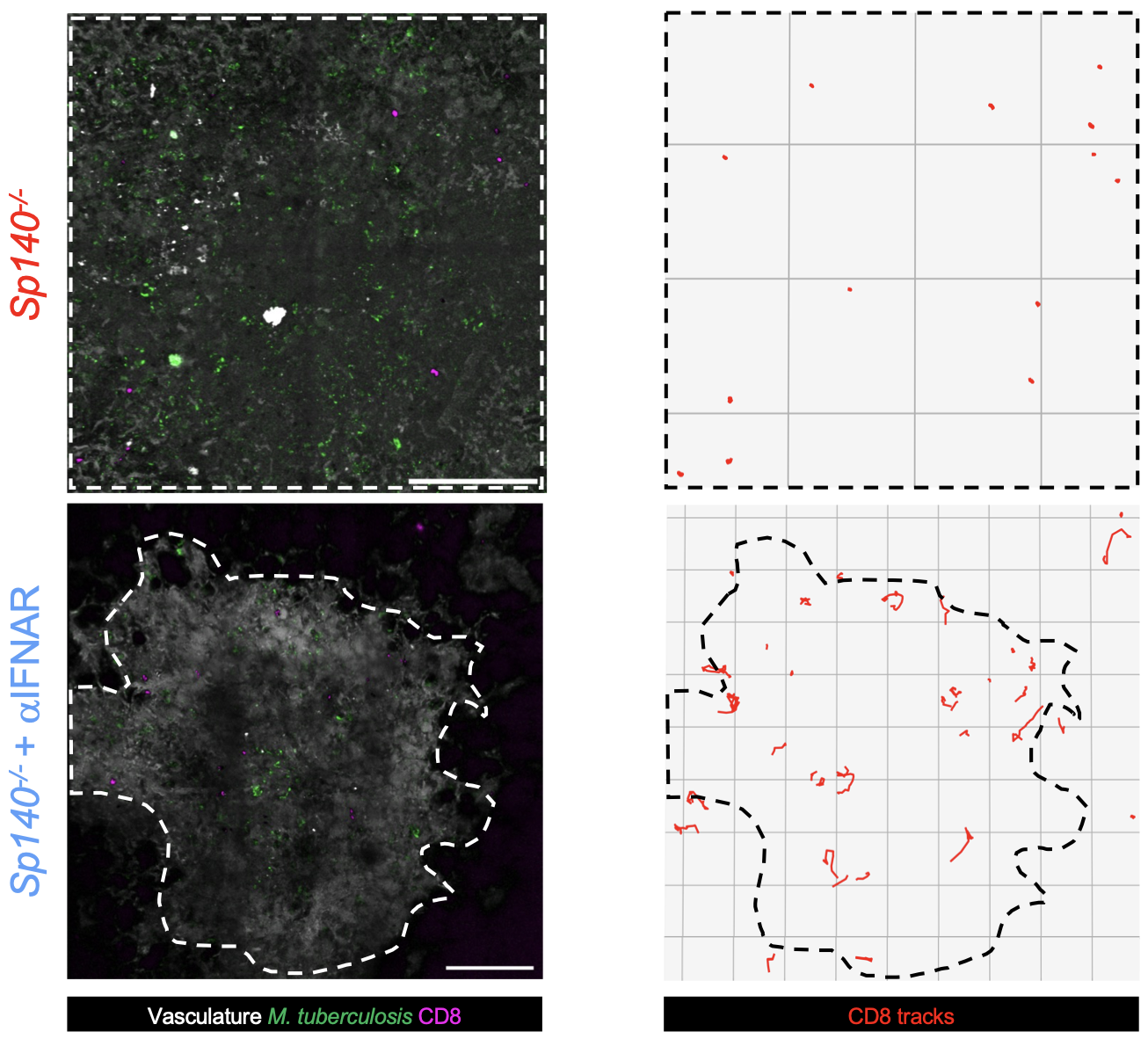
The type I interferon-dominant environment impairs both CD8+ and CD4+ T cell mobility and access to TB lesions. Representative intravital microscopy images of lungs from M. tuberculosis-mTurquoise-infected mice. Bacteria appear in green, transferred CD8+ T cells are in magenta, and lesion boundaries, identified by dextran staining and M. tuberculosis fluorescence, are outlined by a dotted line. Images show Sp140-/- mice (day 21 post infection, top) and anti-IFNAR-treated Sp140-/- mice (day 28 post infection, bottom). The first frame of the video is shown (left), with corresponding CD8+ T cell tracks over the full acquisition period (right). Scale bar, 200 µm. From Caouaille et al. (2026) bioRxiv
More recently, we demonstrated that the chromatin regulator SP140 is essential for maintaining effective CD8⁺ and CD4⁺ T cell immunity. In its absence, excessive type I interferon signaling drives T cell exhaustion, impaired lesion surveillance, and increased bacterial burden—defects that can be reversed by IFNAR blockade.
Together, these findings reveal how immune cell plasticity, metabolic reprogramming, and cytokine balance shape protective immunity in tuberculosis. Looking ahead, we aim to define the molecular circuits that couple metabolism to ILC and T cell function, investigate how type I and type II interferons can be therapeutically balanced, and explore host-directed strategies that enhance protective lymphocyte responses while limiting immunopathology. By integrating single-cell technologies, spatial imaging, and translational models, our goal is to design innovative interventions that strengthen lung immunity and improve TB control.
Deciphering HIV-TB Co-Infection
Principal Investigator | Geanncarlo LUGO
Other Personnel (Alumni) | Maeva DUPONT, Natacha FAIVRE, Claire LASTRUCCI, Sarah MONARD, Shanti SOURIANT, Zoï VAHLAS
Tuberculosis (TB) and acquired immune deficiency syndrome (AIDS) are among the deadliest diseases due to a single infectious agent. Worsening these public health issues is the fact that the two pathogens responsible for these diseases, Mtb and HIV respectively, are frequently associated. The development of new therapeutic strategies requires a better understanding of the synergistic relationship between Mtb and HIV-1, including the consequences of co-infection in the human immune system. Indeed, both pathogens are able to impair the host immune response and have a convergent cellular target in macrophages. Both pathogens alter the macrophage microbicidal functions and convert these cells into cellular reservoirs by modulating their activation process.
While the mechanisms for how HIV-1 render the host susceptible to TB are well-known, those responsible for the TB-driven exacerbation of HIV-1 infection are ill defined. We hypothesize that TB induces important changes in the activation and functional program of human macrophages rendering susceptibility to HIV-1.
In the context of co-infection, a predominant hypothesis to explain exacerbation of HIV-1 infection is that Mtb activates macrophages, which in turn create a “bystander” effect to modulate the local environments through inflammatory signals favoring HIV-1 replication.
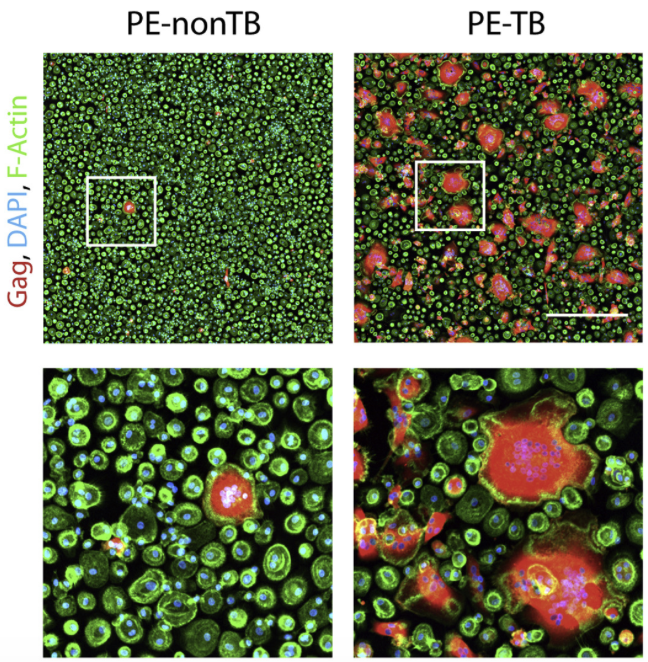
TB-induced microenvironment exacerbates HIV-1 infection of M(IL-10) macrophages. Representative IF images of day 13 HIV-1-infected macrophages treated with PE-nonTB or PE-TB. HIV-1 Gag (red), F-actin (green), and DAPI (blue). Scale bar, 500 μm. Insets are 4× zooms (lower panels). PE, pleural effusion. From Souriant et al. (2019) Cell Rep
In close collaboration with the Verollet lab at IPBS, and in the context of our International Associated Lab with Dr. Luciana Balboa, University of Buenos Aires, Argentina, our work revealed how tuberculosis reshapes immune cell metabolism and signaling to favor both bacterial persistence and viral dissemination. We demonstrated that Mtb reprograms dendritic cells and macrophages through HIF-1α–dependent metabolic pathways: while glycolysis is required for dendritic cell migration, monocytes from TB patients are metabolically biased and fail to generate properly migratory DCs, delaying adaptive immunity. In macrophages, TB-associated microenvironments—particularly lipid mediators—impair HIF-1α–driven glycolytic reprogramming, skewing cells away from microbicidal M1 functions and promoting bacterial survival.
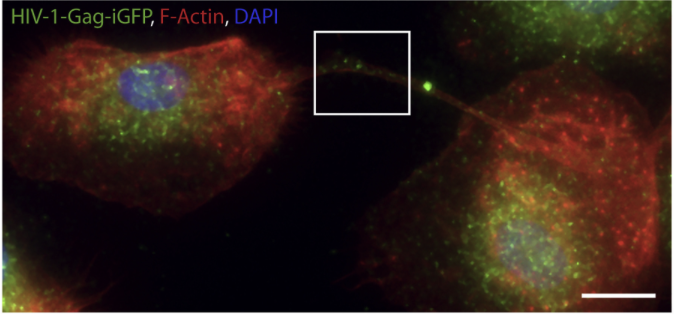
TB enhances HIV-1-induced TNT formation. Immunofluorescence (IF) images showing viral particles in TNT of day 6 HIV-1-Gag-iGFP-infected macrophages previously treated with CmMTB. HIV-1-Gag-GFP (green), F-actin (red), and DAPI (blue). Scale bar, 10 μm. Inset is 4× zoom. From Souriant et al. (2019) Cell Rep
In the context of HIV-1 co-infection, Mtb-induced type I interferon (IFN-I) signaling creates a permissive state for viral replication by rendering macrophages hyporesponsive to antiviral IFN stimulation, driving STAT1-dependent dysfunction, and inducing Siglec-1 expression and IL-10/STAT3–dependent tunneling nanotube (TNT) formation that enhances cell-to-cell viral transfer. Together, these findings position metabolic rewiring and dysregulated IFN-I signaling as central mechanisms linking TB immunopathology to HIV-1 exacerbation, and identify HIF-1α, IFN-I/STAT1, Siglec-1, and TNT pathways as promising targets for host-directed therapies in TB and TB/HIV co-infection.
Stromal Cell Contributions to Granuloma Function and TB Immunity
Principal Investigator | Geanncarlo LUGO
Other Personnel | Elizabeth BAUTISTA, Clara DEYTS, Maxime PINGRET
In addition to immune cells, we are exploring the role of stromal and other non-hematopoietic cells in shaping TB immunity. These structural cells are essential architects of lung tissue integrity and granuloma organization, actively influencing immune cell recruitment, activation, spatial positioning, and the local inflammatory and metabolic milieu.
Our recent work has uncovered a previously unrecognized β3-tubulin (TUBB3)-positive cell population within TB granulomas across multiple species, including mice, guinea pigs, non-human primates, and humans. Although TUBB3 is widely used as a pan-neuronal marker, these cells are distinct from conventional pulmonary resident cells and leukocytes and display an elongated, branched morphology, suggesting a specialized stromal phenotype associated with infection-driven tissue remodeling. Notably, their presence is independent of adaptive immunity and is also observed in other infectious contexts, but not in non-infectious inflammatory conditions such as asthma.
These findings point to an unappreciated stromal component of granuloma biology that may regulate immune cell dynamics, local inflammation, and host-pathogen balance. Moving forward, we aim to define the developmental origin, molecular identity, and functional role of these TUBB3⁺ cells, and to decipher how stromal–immune crosstalk shapes protective versus pathological responses. By integrating spatial imaging, single-cell profiling, and functional perturbation approaches, we seek to uncover novel host-directed strategies that leverage the lung microenvironment to improve TB control and other pulmonary infections.
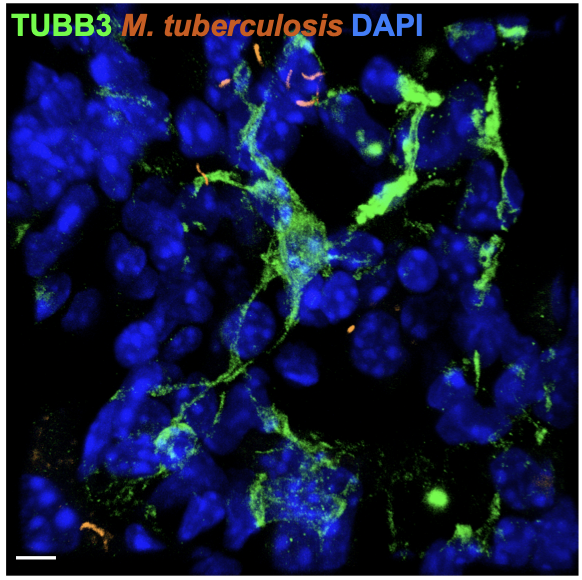
TUBB3+ cells exhibit a branched morphology and are not in a proliferative state. Representative immunohistofluorescence image of lungs from an M. tuberculosis-infected mouse at day 42 post-infection with the H37RV-dsRed strain (orange). TUBB3 (green) and nuclei (DAPI, blue) are shown. Scale bar, 5 µm. From Monard et al. (2025) bioRxiv
Together, our research provides a comprehensive view of tuberculosis pathogenesis, spanning pathogen biology, immune cell function, stromal contributions, and the interplay with HIV co-infection. By integrating molecular, cellular, and in vivo approaches, we uncover how Mtb exploits host pathways, how immune and stromal cells are reprogrammed, and how these processes influence disease outcome. This work highlights critical regulatory circuits, ranging from glycan-lectin interactions and metabolic rewiring to interferon signaling and tissue architecture, that can be targeted to enhance protective immunity, limit pathology, and inform innovative host-directed therapies. Ultimately, our goal is to translate these mechanistic insights into strategies that improve TB control and patient outcomes worldwide.